Fosphenytoin Prescribing Information
Package insert / product label
Generic name: fosphenytoin sodium
Dosage form: injection, solution
Drug class: Hydantoin anticonvulsants
Medically reviewed by Drugs.com. Last updated on Apr 16, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
FOSPHENYTOIN SODIUM INJECTION, USP, for intravenous or intramuscular use
Initial U.S. Approval: 1996
WARNING: CARDIOVASCULAR RISK ASSOCIATED WITH RAPID INFUSION RATES
See full prescribing information for complete boxed warning.
- The rate of intravenous fosphenytoin sodium injection administration should not exceed 150 mg phenytoin sodium equivalents (PE) per minute in adults and 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients because of the risk of severe hypotension and cardiac arrhythmias.
- Careful cardiac monitoring is needed during and after administering intravenous fosphenytoin sodium injection.
- Reduction in rate of administration or discontinuation of dosing may be needed (2.3, 2.4, 5.2).
Recent Major Changes
Warnings and Precautions (5.9) 1/2023
Indications and Usage for Fosphenytoin
Fosphenytoin sodium injection is indicated for the treatment of generalized tonic-clonic status epilepticus and prevention and treatment of seizures occurring during neurosurgery. Fosphenytoin sodium injection can also be substituted, as short-term use, for oral phenytoin. Fosphenytoin sodium injection should be used only when oral phenytoin administration is not possible. (1)
Fosphenytoin Dosage and Administration
- The dose, concentration, and infusion rate of fosphenytoin sodium injection should always be expressed as phenytoin sodium equivalents (PE) (2.1)
- For Status Epilepticus:
- Adult loading dose is 15 to 20 mg PE/kg at a rate of 100 to 150 mg PE/min (2.3)
- Pediatric loading dose is 15 to 20 mg PE/kg at a rate of 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) (2.3)
-
For Non-emergent Loading and Maintenance Dosing:
- Adult loading dose is 10 to 20 mg PE/kg given IV or IM; initial maintenance dose is 4 to 6 mg PE/kg/day in divided doses (2.4)
- Pediatric loading dose is 10 to 15 mg PE/kg at a rate of 1 to 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower); initial maintenance dose is 2 to 4 mg PE/kg every 12 hours at a rate of 1 to 2 mg PE/kg/min (or 100 mg PE/min, whichever is slower) (2.4)
- Intramuscular Administration:
Dosage Forms and Strengths
Contraindications
- Hypersensitivity to fosphenytoin sodium injection, its ingredients, phenytoin, hydantoins (4)
- Sinus bradycardia, sino-atrial block, second and third degree A-V block, and Adams-Stokes syndrome (4)
- A history of prior acute hepatotoxicity attributable to fosphenytoin sodium injection or phenytoin (4, 5.8)
- Coadministration with delavirdine (4)
Warnings and Precautions
- Dosing Errors: Do not confuse the amount of drug to be given in PE with the concentration of the drug in the vial. Ensure the appropriate volume is withdrawn from the vial when preparing for administration. (5.1)
- Withdrawal Precipitated Seizure: May precipitate status epilepticus. Dose reductions or discontinuation should be done gradually. (5.3)
- Serious Dermatologic Reactions: Discontinue at the first sign of a rash, unless clearly not drug-related. If signs or symptoms suggest SJS/TEN, fosphenytoin sodium injection should not be resumed; consider alternative therapy. (5.4)
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity: If signs or symptoms of hypersensitivity are present, evaluate the patient immediately. Discontinue if an alternative etiology cannot be established. (5.5)
- Angioedema: Discontinue immediately if symptoms of angioedema such as facial, perioral, or upper airway swelling occur. (5.7)
- Hematopoietic Complications: If occurs, follow-up observation is indicated and an alternative antiepileptic treatment should be used. (5.9)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥10%) are:
- Adults: pruritus, nystagmus, dizziness, somnolence, and ataxia
- Pediatrics: vomiting, nystagmus, and ataxia (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Hikma Pharmaceuticals USA Inc. at 1-877-845-0689 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
- Pregnancy: Phenytoin (the active metabolite of fosphenytoin sodium injection) prenatal exposure may increase risks for congenital malformations and other adverse developmental outcomes (5.15, 8.1)
- Renal and/or Hepatic Impairment or Hypoalbuminemia: Monitor unbound phenytoin concentrations in these patients (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2024
Related/similar drugs
gabapentin, clonazepam, lamotrigine, lorazepam, pregabalin, topiramate, diazepam
Full Prescribing Information
WARNING: CARDIOVASCULAR RISK ASSOCIATED WITH RAPID INFUSION RATES
The rate of intravenous fosphenytoin sodium injection administration should not exceed 150 mg phenytoin sodium equivalents (PE) per minute in adults and 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients because of the risk of severe hypotension and cardiac arrhythmias. Careful cardiac monitoring is needed during and after administering intravenous fosphenytoin sodium injection. Although the risk of cardiovascular toxicity increases with infusion rates above the recommended infusion rate, these events have also been reported at or below the recommended infusion rate. Reduction in rate of administration or discontinuation of dosing may be needed [see Dosage and Administration (2.3, 2.4) and Warnings and Precautions (5.2)].
1. Indications and Usage for Fosphenytoin
Fosphenytoin sodium injection is indicated for the treatment of generalized tonic-clonic status epilepticus and prevention and treatment of seizures occurring during neurosurgery. Fosphenytoin sodium injection can also be substituted, short-term, for oral phenytoin. Fosphenytoin sodium injection should be used only when oral phenytoin administration is not possible [see Dosage and Administration (2.4) and Warnings and Precautions (5.2)].
2. Fosphenytoin Dosage and Administration
2.1 Important Administration Instructions to Avoid Dosing Errors
Use caution when administering fosphenytoin sodium injection because of the risk of dosing errors [see Warnings and Precautions (5.1)].
Phenytoin Sodium Equivalents (PE)
The dose, concentration, and infusion rate of fosphenytoin sodium injection should always be expressed as phenytoin sodium equivalents (PE). There is no need to perform molecular weight-based adjustments when converting between fosphenytoin and phenytoin sodium doses. Fosphenytoin sodium injection should always be prescribed and dispensed in phenytoin sodium equivalent units (PE). The amount and concentration of fosphenytoin is always expressed in terms of mg of phenytoin sodium equivalents (mg PE).
Concentration of 50 mg PE/mL
Do not confuse the concentration of fosphenytoin sodium injection with the total amount of drug in the vial.
Errors, including fatal overdoses, have occurred when the concentration of the vial (50 mg PE/mL) was misinterpreted to mean that the total content of the vial was 50 mg PE. These errors have resulted in two- or ten-fold overdoses of fosphenytoin sodium injection since each of the vials actually contains a total of 100 mg PE (2 mL vial) or 500 mg PE (10 mL vial). Ensure the appropriate volume of fosphenytoin sodium injection is withdrawn from the vial when preparing the dose for administration. Attention to these details may prevent some fosphenytoin sodium injection medication errors from occurring.
2.2 Preparation
Prior to intravenous (IV) infusion, dilute fosphenytoin sodium injection in 5% Dextrose Injection or 0.9% Sodium Chloride Injection to a concentration ranging from 1.5 to 25 mg PE/mL. The maximum concentration of fosphenytoin sodium injection in any solution should be 25 mg PE/mL. When fosphenytoin sodium injection is given as an IV infusion, fosphenytoin sodium injection needs to be diluted and should only be administered at a rate not exceeding 150 mg PE/min.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
For single-dose only. After opening, any unused product should be discarded.
2.3 Status Epilepticus
- Because of the risk of hypotension and cardiac arrhythmias, the rate of administration for IV fosphenytoin sodium injection should be no greater than 150 mg PE/min in adults and 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients [see Warnings and Precautions (5.2)]. Continuous monitoring of the electrocardiogram, blood pressure, and respiratory function is essential, and the patient should be observed throughout the period where maximal serum phenytoin concentrations occur, approximately 10 to 20 minutes after the end of fosphenytoin sodium injection infusions.
- Because the full antiepileptic effect of phenytoin, whether given as fosphenytoin sodium injection or parenteral phenytoin, is not immediate, other measures, including concomitant administration of an IV benzodiazepine, will usually be necessary for the control of status epilepticus.
- The loading dose should be followed by maintenance doses of either fosphenytoin sodium injection or phenytoin [see Dosage and Administration (2.4)].
- If administration of fosphenytoin sodium injection does not terminate seizures, the use of other anticonvulsants and other appropriate measures should be considered.
Adult and Pediatric Status Epilepticus Dosing:
Table 1. Status Epilepticus Loading Dosages
|
Population |
Dosage |
Infusion rate |
|
Adult |
15 mg PE/kg to 20 mg PE/kg |
100 mg PE/min to 150 mg PE/min, do not exceed a maximum rate of 150 mg PE/min |
|
Pediatric (Birth to less than 17 years of age) |
15 mg PE/kg to 20 mg PE/kg |
2 mg PE/kg/min, or 150 mg PE/min, whichever is slower |
Even though loading doses of fosphenytoin sodium injection have been given by the IM route for other indications when IV access is impossible, IM fosphenytoin sodium injection should ordinarily not be used in the treatment of status epilepticus because therapeutic phenytoin concentrations may not be reached as quickly as with IV administration.
Intramuscular administration of fosphenytoin sodium injection should ordinarily not be used in pediatric patients. When IV access has been impossible, loading doses of fosphenytoin sodium injection have been given by the IM route.
2.4 Non-emergent Loading and Maintenance Dosing
- Because of the risk of hypotension and cardiac arrhythmias, the rate of administration for IV fosphenytoin sodium injection should be no greater than 150 mg PE/min in adults. For loading doses in pediatric patients, the rate should not exceed 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower). For maintenance doses in pediatric patients, the rate should not exceed 1 to 2 mg PE/kg/min (or 100 mg PE/min, whichever is slower). Continuous monitoring of the electrocardiogram, blood pressure, and respiratory function is essential, and the patient should be observed throughout the period where maximal serum phenytoin concentrations occur (approximately 10 to 20 minutes after the end of fosphenytoin sodium injection infusions).
- After the initial maintenance dose, subsequent maintenance doses should be individualized by monitoring serum phenytoin concentrations to achieve a target therapeutic concentration of phenytoin [see Dosage and Administration (2.5) and Warnings and Precautions (5.17)].
Adult and Pediatric Non-emergent Loading and Maintenance Dosing:
Table 2. Non-emergent Loading Dosages
|
Population |
Dosage |
Infusion rate |
|
Adult |
10 mg PE/kg to 20 mg PE/kg |
Not to exceed a maximum rate of 150 mg PE/min |
|
Pediatric (Birth to less than 17 years of age) |
10 mg PE/kg to 15 mg PE/kg |
1 mg PE/kg/min to 2 mg PE/kg/min, or 150 mg PE/min, whichever is slower |
Table 3. Maintenance Dosages
|
Population |
Dosage |
Infusion rate |
|
Adult |
Initial Maintenance Dosage: 4 mg PE/kg/day to 6 mg PE/kg/day in divided doses |
Not to exceed a maximum rate of 150 mg PE/min |
|
Pediatric (Birth to less than 17 years of age) |
Initial Maintenance Dosage: 2 mg PE/kg to 4 mg PE/kg (dose given 12 hours after the loading dose) |
1 mg PE/kg/min to 2 mg PE/kg/min, or 100 mg PE/min, whichever is slower |
|
Maintenance Dosage after Initial Maintenance Dosage: 4 mg PE/kg/day to 8 mg PE/kg/day in divided doses (continued every 12 hours after initial maintenance dose) |
1 mg PE/kg/min to 2 mg PE/kg/min, or 100 mg PE/min, whichever is slower |
Because of the risks of cardiac and local toxicity associated with intravenous fosphenytoin sodium injection, oral phenytoin should be used whenever possible. Intramuscular administration of fosphenytoin sodium injection should ordinarily not be used in pediatric patients.
2.5 Laboratory Tests and Monitoring Levels
Laboratory Tests:
Fosphenytoin sodium injection (or phenytoin) doses are usually selected to attain therapeutic serum total phenytoin concentrations of 10 to 20 mcg/mL (unbound phenytoin concentrations of 1 to 2 mcg/mL). Following fosphenytoin sodium injection administration, it is recommended that phenytoin concentrations not be monitored until conversion to phenytoin is essentially complete. This occurs within approximately 2 hours after the end of IV infusion and 4 hours after intramuscular (IM) injection. Prior to complete conversion, commonly used immunoanalytical techniques, such as TDx®/TDxFLx™ (fluorescence polarization) and Emit® 2000 (enzyme multiplied), may significantly overestimate serum phenytoin concentrations because of cross-reactivity with fosphenytoin. The error is dependent on serum phenytoin and fosphenytoin concentration (influenced by fosphenytoin sodium injection dose, route and rate of administration, and time of sampling relative to dosing), and analytical method. Chromatographic assay methods accurately quantitate phenytoin concentrations in biological fluids in the presence of fosphenytoin. Prior to complete conversion, blood samples for phenytoin monitoring should be collected in tubes containing EDTA as an anticoagulant to minimize ex vivo conversion of fosphenytoin to phenytoin. However, even with specific assay methods, phenytoin concentrations measured before conversion of fosphenytoin is complete will not reflect phenytoin concentrations ultimately achieved.
Monitoring Levels:
Trough levels provide information about clinically effective serum level range and are obtained just prior to the patient's next scheduled dose. Peak levels indicate an individual's threshold for emergence of dose-related side effects and are obtained at the time of expected peak concentration. Therapeutic effect without clinical signs of toxicity occurs more often with serum total phenytoin concentrations between 10 and 20 mcg/mL (unbound phenytoin concentrations of 1 to 2 mcg/mL), although some mild cases of tonic-clonic (grand mal) epilepsy may be controlled with lower serum levels of phenytoin. In patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of unbound phenytoin concentrations may be more relevant [see Dosage and Administration (2.7)].
2.6 Parenteral Substitution for Oral Phenytoin Therapy
When treatment with oral phenytoin is not possible, fosphenytoin sodium injection can be substituted for oral phenytoin at the same total daily phenytoin sodium equivalents (PE) dose. Dilantin capsules are approximately 90% bioavailable by the oral route. Phenytoin, derived from administration of fosphenytoin sodium injection, is 100% bioavailable by both the IM and IV routes. For this reason, serum phenytoin concentrations may increase modestly when IM or IV fosphenytoin sodium injection is substituted for oral phenytoin sodium therapy. The rate of administration for IV fosphenytoin sodium injection should be no greater than 150 mg PE/min in adults and 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients. In controlled trials, IM fosphenytoin sodium injection was administered as a single daily dose utilizing either 1 or 2 injection sites. Some patients may require more frequent dosing. Intramuscular administration of fosphenytoin sodium injection should ordinarily not be used in pediatric patients.
2.7 Dosing in Patients with Renal or Hepatic Impairment or Hypoalbuminemia
Because the fraction of unbound phenytoin (the active metabolite of fosphenytoin sodium injection) is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients. After IV fosphenytoin sodium injection administration to patients with renal and/or hepatic disease, or in those with hypoalbuminemia, fosphenytoin clearance to phenytoin may be increased without a similar increase in phenytoin clearance. This has the potential to increase the frequency and severity of adverse events [see Warnings and Precautions (5.13)].
2.8 Dosing in Geriatrics
The clearance of phenytoin (the active metabolite of fosphenytoin sodium injection) is decreased slightly in elderly patients and lower or less frequent dosing may be required [see Clinical Pharmacology (12.3)].
2.9 Dosing during Pregnancy
Decreased serum concentrations of phenytoin (the active metabolite of fosphenytoin sodium injection) may occur during pregnancy because of altered phenytoin pharmacokinetics [see Clinical Pharmacology (12.3)]. Periodic measurement of serum phenytoin concentrations should be performed during pregnancy, and the fosphenytoin sodium injection dosage should be adjusted as necessary. Postpartum restoration of the original dosage will probably be indicated [see Use in Specific Populations (8.1)]. Because of potential changes in protein binding during pregnancy, the monitoring of phenytoin serum levels should be based on the unbound fraction.
3. Dosage Forms and Strengths
Fosphenytoin sodium injection is a clear, colorless to pale yellow solution available as 50 mg phenytoin sodium equivalents (PE) per mL in:
- 10 mL single-dose injection vials, each containing 500 mg PE/10 mL (50 mg PE/mL)
- 2 mL single-dose injection vials, each containing 100 mg PE/2 mL (50 mg PE/mL)
4. Contraindications
Fosphenytoin sodium injection is contraindicated in patients with:
- A history of hypersensitivity to fosphenytoin sodium injection or its inactive ingredients, or to phenytoin or other hydantoins [see Warnings and Precautions (5.6)]. Reactions have included angioedema.
- Sinus bradycardia, sino-atrial block, second and third degree A-V block, or Adams-Stokes syndrome because of the effect of parenteral phenytoin or fosphenytoin sodium injection on ventricular automaticity.
- A history of prior acute hepatotoxicity attributable to fosphenytoin sodium injection or phenytoin [see Warnings and Precautions (5.8)].
- Coadministration with delavirdine because of the potential for loss of virologic response and possible resistance to delavirdine or to the class of non-nucleoside reverse transcriptase inhibitors.
5. Warnings and Precautions
5.1 Dosing Errors
Phenytoin Sodium Equivalents (PE)
Do not confuse the amount of drug to be given in PE with the concentration of the drug in the vial.
Doses of fosphenytoin sodium injection are always expressed in terms of milligrams of phenytoin sodium equivalents (mg PE). 1 mg PE is equivalent to 1 mg phenytoin sodium.
Do not, therefore, make any adjustment in the recommended doses when substituting fosphenytoin sodium injection for phenytoin sodium or vice versa. For example, if a patient is receiving 1000 mg PE of fosphenytoin sodium injection, that is equivalent to 1000 mg of phenytoin sodium.
Concentration of 50 mg PE/mL
Medication errors associated with fosphenytoin sodium injection have resulted in patients receiving the wrong dose of fosphenytoin. Fosphenytoin sodium injection is marketed in 2 mL vials containing a total of 100 mg PE and 10 mL vials containing a total of 500 mg PE. The concentration of each vial is 50 mg PE/mL. Errors have occurred when the concentration of the vial (50 mg PE/mL) was misinterpreted to mean that the total content of the vial was 50 mg PE. These errors have resulted in two- or ten-fold overdoses of fosphenytoin sodium injection since each vial actually contains a total of 100 mg PE or 500 mg PE. In some cases, ten-fold overdoses were associated with fatal outcomes. To help minimize confusion, the prescribed dose of fosphenytoin sodium injection should always be expressed in milligrams of phenytoin equivalents (mg PE) [see Dosage and Administration (2.1)]. Additionally, when ordering and storing fosphenytoin sodium injection, consider displaying the total drug content (i.e., 100 mg PE/2 mL or 500 mg PE/10 mL) instead of concentration in computer systems, pre-printed orders, and automated dispensing cabinet databases to help ensure that total drug content can be clearly identified. Care should be taken to ensure the appropriate volume of fosphenytoin sodium injection is withdrawn from the vial when preparing the drug for administration. Attention to these details may prevent some fosphenytoin sodium injection medication errors from occurring.
5.2 Cardiovascular Risk Associated with Rapid Infusion
Rapid intravenous administration of fosphenytoin sodium injection increases the risk of adverse cardiovascular reactions, including severe hypotension and cardiac arrhythmias. Cardiac arrhythmias have included bradycardia, heart block, QT interval prolongation, ventricular tachycardia, and ventricular fibrillation which have resulted in asystole, cardiac arrest, and death. Severe complications are most commonly encountered in critically ill patients, elderly patients, and patients with hypotension and severe myocardial insufficiency. However, cardiac events have also been reported in adults and children without underlying cardiac disease or comorbidities and at recommended doses and infusion rates.
The rate of intravenous fosphenytoin sodium injection administration should not exceed 150 mg phenytoin sodium equivalents (PE) per minute in adults and 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients [see Dosage and Administration (2.3, 2.4)].
Although the risk of cardiovascular toxicity increases with infusion rates above the recommended infusion rate, these events have also been reported at or below the recommended infusion rate.
As non-emergency therapy, intravenous fosphenytoin sodium injection should be administered more slowly. Because of the risks of cardiac and local toxicity associated with IV fosphenytoin sodium injection, oral phenytoin should be used whenever possible.
Because adverse cardiovascular reactions have occurred during and after infusions, careful cardiac and respiratory monitoring is needed during and after the administration of intravenous fosphenytoin sodium injection. Reduction in rate of administration or discontinuation of dosing may be needed.
5.3 Withdrawal Precipitated Seizure, Status Epilepticus
Antiepileptic drugs should not be abruptly discontinued because of the possibility of increased seizure frequency, including status epilepticus. When, in the judgment of the clinician, the need for dosage reduction, discontinuation, or substitution of alternative antiepileptic medication arises, this should be done gradually. However, in the event of an allergic or hypersensitivity reaction, rapid substitution of alternative therapy may be necessary. In this case, alternative therapy should be an antiepileptic drug not belonging to the hydantoin chemical class.
5.4 Serious Dermatologic Reactions
Fosphenytoin sodium injection can cause severe cutaneous adverse reactions (SCARs), which may be fatal. Reported reactions in phenytoin (the active metabolite of fosphenytoin sodium injection)-treated patients have included toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), acute generalized exanthematous pustulosis (AGEP), and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see Warnings and Precautions (5.5)]. The onset of symptoms is usually within 28 days, but can occur later. Fosphenytoin sodium injection should be discontinued at the first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest a severe cutaneous adverse reaction, use of this drug should not be resumed and alternative therapy should be considered. If a rash occurs, the patient should be evaluated for signs and symptoms of SCARs.
Studies in patients of Chinese ancestry have found a strong association between the risk of developing SJS/TEN and the presence of HLA-B*1502, an inherited allelic variant of the HLA B gene, in patients using carbamazepine. Limited evidence suggests that HLA-B*1502 may be a risk factor for the development of SJS/TEN in patients of Asian ancestry taking other antiepileptic drugs associated with SJS/TEN, including phenytoin. In addition, retrospective, case-control, genome-wide association studies in patients of southeast Asian ancestry have also identified an increased risk of SCARs in carriers of the decreased function CYP2C9*3 variant, which has also been associated with decreased clearance of phenytoin. Consider avoiding fosphenytoin sodium injection as an alternative to carbamazepine in patients who are positive for HLA-B*1502 or in CYP2C9*3 carriers.
Should fosphenytoin sodium injection be utilized for CYP2C9*3 carriers, consider starting at the lower end of the dosage range [see Use in Specific Populations (8.7)].
The use of HLA-B*1502 or CYP2C9 genotyping has important limitations and must never substitute for appropriate clinical vigilance and patient management. The role of other possible factors in the development of, and morbidity from, SJS/TEN, such as antiepileptic drug (AED) dose, compliance, concomitant medications, comorbidities, and the level of dermatologic monitoring have not been studied.
5.5 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan hypersensitivity, has been reported in patients taking antiepileptic drugs, including phenytoin and fosphenytoin sodium injection.
Some of these events have been fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Fosphenytoin sodium injection should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
5.6 Hypersensitivity
Fosphenytoin sodium injection and other hydantoins are contraindicated in patients who have experienced phenytoin hypersensitivity [see Contraindications (4) and Warnings and Precautions (5.7)]. Additionally, consider alternatives to structurally similar drugs such as carboxamides (e.g., carbamazepine), barbiturates, succinimides, and oxazolidinediones (e.g., trimethadione) in these same patients. Similarly, if there is a history of hypersensitivity reactions to these structurally similar drugs in the patient or immediate family members, consider alternatives to fosphenytoin sodium injection.
5.7 Angioedema
Angioedema has been reported in patients treated with phenytoin and fosphenytoin sodium injection in the postmarketing setting. Fosphenytoin sodium injection should be discontinued immediately if symptoms of angioedema, such as facial, perioral, or upper airway swelling occur. Fosphenytoin sodium injection should be discontinued permanently if a clear alternative etiology for the reaction cannot be established.
5.8 Hepatic Injury
Cases of acute hepatotoxicity, including infrequent cases of acute hepatic failure, have been reported with phenytoin (the active metabolite of fosphenytoin sodium injection). These events may be part of the spectrum of DRESS or may occur in isolation [see Warnings and Precautions (5.5)]. Other common manifestations include jaundice, hepatomegaly, elevated serum transaminase levels, leukocytosis, and eosinophilia. The clinical course of acute phenytoin hepatotoxicity ranges from prompt recovery to fatal outcomes. In these patients with acute hepatotoxicity, fosphenytoin sodium injection should be immediately discontinued and not re-administered.
5.9 Hematopoietic Complications
Hematopoietic complications, some fatal, have occasionally been reported in association with administration of phenytoin (the active metabolite of fosphenytoin sodium injection). These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression.
There have been a number of reports that have suggested a relationship between phenytoin and the development of lymphadenopathy (local or generalized), including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin's disease. Although a cause and effect relationship has not been established, the occurrence of lymphadenopathy indicates the need to differentiate such a condition from other types of lymph node pathology. Lymph node involvement may occur with or without symptoms and signs resembling DRESS [see Warnings and Precautions (5.5)].
In all cases of lymphadenopathy, follow-up observation for an extended period is indicated and every effort should be made to achieve seizure control using alternative antiepileptic drugs. Macrocytosis and megaloblastic anemia have occurred, these conditions usually respond to folic acid therapy. Pure red cell aplasia has also been reported with phenytoin.
5.10 Sensory Disturbances
Severe burning, itching, and/or paresthesia were reported by 7 of 16 normal volunteers administered IV fosphenytoin sodium injection at a dose of 1200 mg PE at the maximum rate of administration (150 mg PE/min). The severe sensory disturbance lasted from 3 to 50 minutes in 6 of these subjects and for 14 hours in the seventh subject. In some cases, milder sensory disturbances persisted for as long as 24 hours. The location of the discomfort varied among subjects with the groin mentioned most frequently as an area of discomfort. In a separate cohort of 16 normal volunteers (taken from 2 other studies) who were administered IV fosphenytoin sodium injection at a dose of 1200 mg PE at the maximum rate of administration (150 mg PE/min), none experienced severe disturbances, but most experienced mild to moderate itching or tingling. Patients administered fosphenytoin sodium injection at doses of 20 mg PE/kg at 150 mg PE/min are expected to experience discomfort of some degree. The occurrence and intensity of the discomfort can be lessened by slowing or temporarily stopping the infusion. The effect of continuing infusion unaltered in the presence of these sensations is unknown. No permanent sequelae have been reported thus far. The pharmacologic basis for these positive sensory phenomena is unknown, but other phosphate ester drugs, which deliver smaller phosphate loads, have been associated with burning, itching, and/or tingling predominantly in the groin area.
5.11 Local Toxicity (Including Purple Glove Syndrome)
Edema, discoloration, and pain distal to the site of injection (described as "purple glove syndrome") have also been reported following peripheral intravenous fosphenytoin sodium injection. This may or may not be associated with extravasation. The syndrome may not develop for several days after injection.
5.12 Phosphate Load
The phosphate load provided by fosphenytoin sodium injection (0.0037 mmol phosphate/mg PE fosphenytoin sodium injection) should be considered when treating patients who require phosphate restriction, such as those with severe renal impairment.
5.13 Renal or Hepatic Disease or Hypoalbuminemia
Because the fraction of unbound phenytoin (the active metabolite of fosphenytoin sodium injection) is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients. After IV administration to patients with renal and/or hepatic disease, or in those with hypoalbuminemia, fosphenytoin clearance to phenytoin may be increased without a similar increase in phenytoin clearance. This has the potential to increase the frequency and severity of adverse events.
5.14 Exacerbation of Porphyria
In view of isolated reports associating phenytoin (the active metabolite of fosphenytoin sodium injection) with exacerbation of porphyria, caution should be exercised in using fosphenytoin sodium injection in patients suffering from this disease.
5.15 Teratogenicity and Other Harm to the Newborn
Fosphenytoin sodium injection may cause fetal harm when administered to a pregnant woman. Prenatal exposure to phenytoin (the active metabolite of fosphenytoin sodium injection) may increase the risks for congenital malformations and other adverse developmental outcomes [see Use in Specific Populations (8.1)].
Increased frequencies of major malformations (such as orofacial clefts and cardiac defects), and abnormalities characteristic of fetal hydantoin syndrome, including dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits, have been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy. There have been several reported cases of malignancies, including neuroblastoma.
A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.
5.16 Hyperglycemia
Hyperglycemia, resulting from the inhibitory effect of phenytoin (the active metabolite of fosphenytoin sodium injection) on insulin release, has been reported. Phenytoin may also raise the serum glucose concentrations in diabetic patients.
5.17 Serum Phenytoin Levels above Therapeutic Range
Serum levels of phenytoin (the active metabolite of fosphenytoin sodium injection) sustained above the therapeutic range may produce confusional states referred to as "delirium," "psychosis," or "encephalopathy," or rarely, irreversible cerebellar dysfunction and/or cerebellar atrophy. Accordingly, at the first sign of acute toxicity, serum levels should be immediately checked. Fosphenytoin sodium injection dose reduction is indicated if serum levels are excessive; if symptoms persist, administration of fosphenytoin sodium injection should be discontinued.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- Cardiovascular Risk Associated with Rapid Infusion [see Warnings and Precautions (5.2)]
- Withdrawal Precipitated Seizure, Status Epilepticus [see Warnings and Precautions (5.3)]
- Serious Dermatologic Reactions [see Warnings and Precautions (5.4)]
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity [see Warnings and Precautions (5.5)]
- Hypersensitivity [see Warnings and Precautions (5.6)]
- Angioedema [see Warnings and Precautions (5.7)]
- Hepatic Injury [see Warnings and Precautions (5.8)]
- Hematopoietic Complications [see Warnings and Precautions (5.9)]
- Sensory Disturbances [see Warnings and Precautions (5.10)]
- Local Toxicity (Including Purple Glove Syndrome) [see Warnings and Precautions (5.11)]
- Exacerbation of Porphyria [see Warnings and Precautions (5.14)]
- Teratogenicity and Other Harm to the Newborn [see Warnings and Precautions (5.15)]
- Hyperglycemia [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The more important adverse clinical reactions caused by the IV use of fosphenytoin sodium injection or phenytoin are cardiovascular collapse and/or CNS depression. Hypotension can occur when either drug is administered rapidly by the IV route. The rate of administration is very important; for fosphenytoin sodium injection, it should not exceed 150 mg PE/min [see Warnings and Precautions (5.2)]. The adverse reactions most commonly observed with the use of fosphenytoin sodium injection in clinical trials were nystagmus, dizziness, pruritus, somnolence, and ataxia. With one exception, these reactions are commonly associated with the administration of IV phenytoin. Pruritus, however, was seen much more often following fosphenytoin sodium injection administration and occurred more often with IV fosphenytoin sodium injection administration than with IM fosphenytoin sodium injection administration. These reactions were dose and rate related; most alert patients (41 of 64; 64%) administered doses of ≥15 mg PE/kg at 150 mg PE/min experienced discomfort of some degree. These sensations, generally described as itching, burning, or tingling, were usually not at the infusion site. The location of the discomfort varied with the groin mentioned most frequently as a site of involvement. The paresthesia and pruritus were transient events that occurred within several minutes of the start of infusion and generally resolved within 10 minutes after completion of fosphenytoin sodium injection infusion. Some patients experienced symptoms for hours. These reactions did not increase in severity with repeated administration. Concurrent adverse events or clinical laboratory change suggesting an allergic process were not seen [see Warnings and Precautions (5.10)]. Approximately 2% of the 859 patients who received fosphenytoin sodium injection in premarketing clinical trials discontinued treatment because of an adverse event. The adverse events most commonly associated with withdrawal were pruritus (0.5%), hypotension (0.3%), and bradycardia (0.2%).
Dose and Rate Dependency of Adverse Reactions Following IV Fosphenytoin Sodium Injection: The incidence of adverse reactions tended to increase as both dose and infusion rate increased. In particular, at doses of ≥15mg PE/kg and rates ≥150 mg PE/min, transient pruritus, tinnitus, nystagmus, somnolence, and ataxia occurred 2 to 3 times more often than at lower doses or rates.
Incidence in Controlled Clinical Trials
All adverse events were recorded during the trials by the clinical investigators using terminology of their own choosing. Similar types of events were grouped into standardized categories using modified COSTART dictionary terminology. These categories are used in the tables and listings below with the frequencies representing the proportion of individuals exposed to fosphenytoin sodium injection or comparative therapy.
Incidence in Controlled Clinical Trials - IV Administration to Adult Patients with Epilepsy or Neurosurgical Patients: Table 4 lists adverse reactions that occurred in at least 2% of patients treated with IV fosphenytoin sodium injection at the maximum dose and rate in a randomized, double-blind, controlled clinical trial where the rates for phenytoin and fosphenytoin sodium injection administration would have resulted in equivalent systemic exposure to phenytoin.
TABLE 4. Adverse Reaction Incidence Following IV Administration at the Maximum Dose and Rate to Adult Patients with Epilepsy or Neurosurgical Patients (Events in at Least 2% of Fosphenytoin Sodium Injection-Treated Patients)
|
BODY SYSTEM Adverse Event |
IV Fosphenytoin Sodium Injection N=90 |
IV Phenytoin1 N=22 |
|
BODY AS A WHOLE | ||
|
Pelvic Pain |
4 |
0 |
|
Asthenia |
2 |
0 |
|
Back Pain |
2 |
0 |
|
Headache |
2 |
5 |
|
CARDIOVASCULAR | ||
|
Hypotension |
8 |
9 |
|
Vasodilatation |
6 |
5 |
|
Tachycardia |
2 |
0 |
|
DIGESTIVE | ||
|
Nausea |
9 |
14 |
|
Tongue Disorder |
4 |
0 |
|
Dry Mouth |
4 |
5 |
|
Vomiting |
2 |
9 |
|
NERVOUS | ||
|
Nystagmus |
44 |
59 |
|
Dizziness |
31 |
27 |
|
Somnolence |
20 |
27 |
|
Ataxia |
11 |
18 |
|
Stupor |
8 |
5 |
|
Incoordination |
4 |
5 |
|
Paresthesia |
4 |
0 |
|
Extrapyramidal Syndrome |
4 |
0 |
|
Tremor |
3 |
9 |
|
Agitation |
3 |
0 |
|
Hypesthesia |
2 |
9 |
|
Dysarthria |
2 |
0 |
|
Vertigo |
2 |
0 |
|
Brain Edema |
2 |
5 |
|
SKIN AND APPENDAGES | ||
|
Pruritus |
49 |
5 |
|
SPECIAL SENSES | ||
|
Tinnitus |
9 |
9 |
|
Diplopia |
3 |
0 |
|
Taste Perversion |
3 |
0 |
|
Amblyopia |
2 |
9 |
|
Deafness |
2 |
0 |
1 The study was not designed to assess comparative safety.
Incidence in Clinical Trials - IV Administration to Pediatric Patients with Epilepsy or Neurosurgical Patients: The overall incidence of adverse reactions and the types of adverse reactions seen were similar among children and adults treated with fosphenytoin sodium injection. In an open-label, safety, tolerability, and pharmacokinetic study of fosphenytoin in pediatric subjects (neonates through age 16), the following adverse reactions occurred at a frequency of at least 5% in 96 subjects treated with intravenous fosphenytoin sodium injection: vomiting (21%), nystagmus (18%), ataxia (10%), fever (8%), nervousness (7%), pruritus (6%), somnolence (6%), hypotension (5%), and rash (5%).
Incidence in Controlled Trials - IM Administration to Adult Patients with Epilepsy: Table 5 lists adverse reactions that occurred in at least 2% of fosphenytoin sodium injection-treated patients in a double-blind, randomized, controlled clinical trial of adult epilepsy patients receiving either IM fosphenytoin sodium injection substituted for oral phenytoin or continuing oral phenytoin. Both treatments were administered for 5 days.
TABLE 5. Adverse Reaction Incidence Following Substitution of IM Fosphenytoin Sodium Injection for Oral Phenytoin in Adult Patients with Epilepsy (Events in at Least 2% of Fosphenytoin Sodium Injection-Treated Patients)
|
BODY SYSTEM Adverse Event |
IM Fosphenytoin Sodium Injection N=179 |
Oral Phenytoin1 N=61 |
|
BODY AS A WHOLE | ||
|
Headache |
9 |
5 |
|
Asthenia |
9 |
3 |
|
DIGESTIVE | ||
|
Nausea |
5 |
0 |
|
Vomiting |
3 |
0 |
|
HEMATOLOGIC AND LYMPHATIC | ||
|
Ecchymosis |
7 |
5 |
|
NERVOUS | ||
|
Nystagmus |
15 |
8 |
|
Tremor |
10 |
13 |
|
Ataxia |
8 |
8 |
|
Incoordination |
8 |
5 |
|
Somnolence |
7 |
10 |
|
Dizziness |
5 |
3 |
|
Paresthesia |
4 |
3 |
|
Reflexes Decreased |
3 |
5 |
|
SKIN AND APPENDAGES | ||
|
Pruritus |
3 |
0 |
1The study was not designed to assess comparative safety.
Adverse Events During Clinical Trials in Adult and Pediatric Patients
Fosphenytoin sodium injection has been administered to approximately 900 individuals during clinical trials. Adverse events seen at least twice are listed in the following, except those already included in previous tables and listings. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in greater than 1/100 individuals; infrequent adverse events are those occurring in 1/100 to 1/1000 individuals.
Body as a Whole: Frequent: fever, injection-site reaction, infection, chills, face edema, injection-site pain; Infrequent: sepsis, injection-site inflammation, injection-site edema, injection-site hemorrhage, flu syndrome, malaise, generalized edema, shock, photosensitivity reaction, cachexia, cryptococcosis.
Cardiovascular: Frequent: hypertension; Infrequent: cardiac arrest, migraine, syncope, cerebral hemorrhage, palpitation, sinus bradycardia, atrial flutter, bundle branch block, cardiomegaly, cerebral infarct, postural hypotension, pulmonary embolus, QT interval prolongation, thrombophlebitis, ventricular extrasystoles, congestive heart failure.
Digestive: Frequent: constipation; Infrequent: dyspepsia, diarrhea, anorexia, gastrointestinal hemorrhage, increased salivation, liver function tests abnormal, tenesmus, tongue edema, dysphagia, flatulence, gastritis, ileus.
Endocrine: Infrequent: diabetes insipidus.
Hematologic and Lymphatic: Infrequent: thrombocytopenia, anemia, leukocytosis, cyanosis, hypochromic anemia, leukopenia, lymphadenopathy, petechia. [see Warnings and Precautions (5.9)].
Laboratory Test Abnormality: Phenytoin (the active metabolite of fosphenytoin sodium injection) may cause increased serum levels of glucose and alkaline phosphatase.
Metabolic and Nutritional: Frequent: hypokalemia; Infrequent: hyperglycemia, hypophosphatemia, alkalosis, acidosis, dehydration, hyperkalemia, ketosis.
Musculoskeletal: Frequent: myasthenia; Infrequent: myopathy, leg cramps, arthralgia, myalgia.
Nervous: Frequent: reflexes increased, speech disorder, dysarthria, intracranial hypertension, thinking abnormal, nervousness; Infrequent: confusion, twitching, Babinski sign positive, circumoral paresthesia, hemiplegia, hypotonia, convulsion, extrapyramidal syndrome, insomnia, meningitis, depersonalization, CNS depression, depression, hypokinesia, hyperkinesia, paralysis, psychosis, aphasia, emotional lability, coma, hyperesthesia, myoclonus, personality disorder, acute brain syndrome, encephalitis, subdural hematoma, encephalopathy, hostility, akathisia, amnesia, neurosis.
Respiratory: Frequent: pneumonia; Infrequent: pharyngitis, sinusitis, hyperventilation, rhinitis, apnea, aspiration pneumonia, asthma, dyspnea, atelectasis, cough increased, sputum increased, epistaxis, hypoxia, pneumothorax, hemoptysis, bronchitis.
Skin and Appendages: Frequent: rash; Infrequent: maculopapular rash, urticaria, sweating, skin discoloration, contact dermatitis, pustular rash, skin nodule.
Special Senses: Infrequent: visual field defect, eye pain, conjunctivitis, photophobia, hyperacusis, mydriasis, parosmia, ear pain, taste loss.
Urogenital: Infrequent: urinary retention, oliguria, dysuria, vaginitis, albuminuria, genital edema, kidney failure, polyuria, urethral pain, urinary incontinence, vaginal moniliasis.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of phenytoin or fosphenytoin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: Anaphylaxis, angioedema [see Warnings and Precautions (5.7)]
Hematologic:Pure red cell aplasia [see Warnings and Precautions (5.9)]
Laboratory Test Abnormality: Phenytoin or fosphenytoin sodium injection may decrease serum concentrations of T4. It may also produce lower than normal values for dexamethasone or metyrapone tests. Phenytoin may also cause increased serum levels of gamma glutamyl transpeptidase (GGT).
Nervous System Disorders: Dyskinesia
7. Drug Interactions
Fosphenytoin is extensively bound to human plasma proteins. Drugs highly bound to albumin could increase the unbound fraction of fosphenytoin. Although, it is unknown whether this could result in clinically significant effects, caution is advised when administering fosphenytoin sodium injection with other drugs that significantly bind to serum albumin. The most significant drug interactions following administration of fosphenytoin sodium injection are expected to occur with drugs that interact with phenytoin. Phenytoin is extensively bound to serum plasma proteins and is prone to competitive displacement. Phenytoin is primarily metabolized by the hepatic cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19 and is particularly susceptible to inhibitory drug interactions because it is subject to saturable metabolism. Inhibition of metabolism may produce significant increases in circulating phenytoin concentrations and enhance the risk of drug toxicity. Monitoring of phenytoin serum levels is recommended when a drug interaction is suspected.
Phenytoin or fosphenytoin sodium injection is a potent inducer of hepatic drug-metabolizing enzymes.
7.1 Drugs that Affect Phenytoin or Fosphenytoin Sodium Injection
Table 6 includes commonly occurring drug interactions that affect phenytoin (the active metabolite of fosphenytoin sodium injection) concentrations. However, this list is not intended to be inclusive or comprehensive. Individual prescribing information from relevant drugs should be consulted.
The addition or withdrawal of these agents in patients on phenytoin therapy may require an adjustment of the phenytoin dose to achieve optimal clinical outcome.
Table 6. Drugs That Affect Phenytoin Concentrations
| Interacting Agent | Examples |
| Drugs that may increase phenytoin serum levels |
|
| Antiepileptic drugs | Ethosuximide, felbamate, oxcarbazepine, methsuximide, topiramate |
| Azoles | Fluconazole, ketoconazole, itraconazole, miconazole, voriconazole |
| Antineoplastic agents | Capecitabine, fluorouracil |
| Antidepressants | Fluoxetine, fluvoxamine, sertraline |
| Gastric acid reducing agents | H2 antagonists (cimetidine), omeprazole |
| Sulfonamides | Sulfamethizole, sulfaphenazole, sulfadiazine, sulfamethoxazole-trimethoprim |
| Other | Acute alcohol intake, amiodarone, chloramphenicol, chlordiazepoxide, disulfiram, estrogen, fluvastatin, isoniazid, methylphenidate, phenothiazines, salicylates, ticlopidine, tolbutamide, trazodone, warfarin |
| Drugs that may decrease phenytoin serum levels |
|
| Antineoplastic agents usually in combination | Bleomycin, carboplatin, cisplatin, doxorubicin, methotrexate |
| Antiviral agents | Fosamprenavir, nelfinavir, ritonavir |
| Antiepileptic drugs | Carbamazepine, vigabatrin |
| Other | Chronic alcohol abuse, diazepam, diazoxide, folic acid, reserpine, rifampin, St. John's wort,a theophylline |
| Drugs that may either increase or decrease phenytoin serum levels |
|
| Antiepileptic drugs | Phenobarbital, valproate sodium, valproic acid |
a The induction potency of St. John's wort may vary widely based on preparation.
7.2 Drugs Affected by Phenytoin or Fosphenytoin Sodium Injection
Table 7 includes commonly occurring drug interactions affected by phenytoin (the active metabolite of fosphenytoin sodium injection). However, this list is not intended to be inclusive or comprehensive. Individual drug package inserts should be consulted. The addition or withdrawal of phenytoin during concomitant therapy with these agents may require adjustment of the dose of these agents to achieve optimal clinical outcome.
Table 7: Drugs Affected by Phenytoin
|
Interacting Agent |
Examples |
|
Drugs whose efficacy is impaired by phenytoin |
|
|
Azoles |
Fluconazole, ketoconazole, itraconazole, posaconazole, voriconazole |
|
Antineoplastic agents |
Irinotecan, paclitaxel, teniposide |
|
Delavirdine |
Phenytoin can substantially reduce the concentrations of delavirdine. This can lead to loss of virologic response and possible resistance [see Contraindications (4)]. |
|
Neuromuscular blocking agents |
Cisatracurium, pancuronium, rocuronium and vecuronium: resistance to the neuromuscular blocking action of the nondepolarizing neuromuscular blocking agents has occurred in patients chronically administered phenytoin. Whether or not phenytoin has the same effect on other non-depolarizing agents is unknown. Prevention or Management: Patients should be monitored closely for more rapid recovery from neuromuscular blockade than expected, and infusion rate requirements may be higher. |
|
Warfarin |
Increased and decreased PT/INR responses have been reported when phenytoin is coadministered with warfarin. |
|
Other |
Corticosteroids, doxycycline, estrogens, furosemide, oral contraceptives, paroxetine, quinidine, rifampin, sertraline, theophylline, and vitamin D |
|
Drugs whose level is decreased by phenytoin |
|
|
Anticoagulants | Apixaban, dabigatran, edoxaban, rivaroxaban |
|
Antiepileptic drugsa |
Carbamazepine, felbamate, lamotrigine, topiramate, oxcarbazepine, lacosamide |
|
Antilipidemic agents |
Atorvastatin, fluvastatin, simvastatin |
|
Antiplatelets | Ticagrelor |
|
Antiviral agents |
Efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir |
|
Calcium channel blockers |
Nifedipine, nimodipine, nisoldipine, verapamil |
|
Other |
Albendazole (decreases active metabolite), chlorpropamide, clozapine, cyclosporine, digoxin, disopyramide, folic acid, methadone, mexiletine, praziquantel, quetiapine |
aThe effect of phenytoin on phenobarbital, valproic acid and sodium valproate serum levels is unpredictable.
7.3 Hyperammonemia with Concomitant Use of Valproate
Concomitant administration of phenytoin and valproate has been associated with an increased risk of valproate-associated hyperammonemia. Patients treated concomitantly with these two drugs should be monitored for signs and symptoms of hyperammonemia.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as fosphenytoin sodium injection, during pregnancy. Physicians are advised to recommend that pregnant patients taking fosphenytoin sodium injection enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the toll free number 1-888-233-2334, and must be done by patients themselves. Information on the registry can also be found at the website http://www.aedpregnancyregistry.org/.
Risk Summary
In humans, prenatal exposure to phenytoin (the active metabolite of fosphenytoin sodium injection) may increase the risks for congenital malformations and other adverse developmental outcomes. Prenatal phenytoin exposure is associated with an increased incidence of major malformations, including orofacial clefts and cardiac defects. In addition, the fetal hydantoin syndrome, a pattern of abnormalities including dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits has been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy [see Data]. There have been several reported cases of malignancies, including neuroblastoma, in children whose mothers received phenytoin during pregnancy.
Administration of phenytoin to pregnant animals resulted in an increased incidence of fetal malformations and other manifestations of developmental toxicity (including embryofetal death, growth impairment, and behavioral abnormalities) in multiple species at clinically relevant doses [see Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Clinical Considerations
Disease-associated maternal risk
An increase in seizure frequency may occur during pregnancy because of altered phenytoin pharmacokinetics. Periodic measurement of serum phenytoin concentrations may be valuable in the management of pregnant women as a guide to appropriate adjustment of dosage [see Dosage and Administration (2.5, 2.9)]. However, postpartum restoration of the original dosage will probably be indicated.
Fetal/Neonatal adverse reactions
A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.
Data
Human Data
Meta-analyses using data from published observational studies and registries have estimated an approximately 2.4-fold increased risk for any major
malformation in children with prenatal phenytoin exposure compared to controls. An increased risk of heart defects, facial clefts, and digital hypoplasia has been reported. The fetal hydantoin syndrome is a pattern of congenital anomalies including craniofacial anomalies, nail and digital hypoplasia, prenatal-onset growth deficiency, and neurodevelopmental deficiencies.
Animal Data
Administration of phenytoin to pregnant rats, rabbits, and mice during organogenesis resulted in embryofetal death, fetal malformations, and decreased fetal growth. Malformations (including craniofacial, cardiovascular, neural, limb, and digit abnormalities) were observed in rats, rabbits, and mice at doses as low as 100, 75, and 12.5 mg/kg, respectively.
8.2 Lactation
Risk Summary
It is not known whether fosphenytoin is secreted in human milk. Following administration of phenytoin (the active metabolite of fosphenytoin sodium injection), phenytoin is secreted in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for fosphenytoin sodium injection and any potential adverse effects on the breastfed infant from fosphenytoin sodium injection or from the underlying maternal condition.
8.4 Pediatric Use
Fosphenytoin sodium injection is indicated for the treatment of generalized tonic-clonic status epilepticus and prevention and treatment of seizures occurring during neurosurgery in all pediatric age groups [see Indications and Usage (1) and Dosage and Administration (2.3, 2.4)]. Because rapid intravenous administration of fosphenytoin sodium injection increases the risk of adverse cardiovascular reactions, the rate of administration should not exceed 2 mg PE/kg/min (or 150 mg PE/min, whichever is slower) in pediatric patients [see Dosage and Administration (2.3, 2.4) and Warnings and Precautions (5.2)].
8.5 Geriatric Use
No systematic studies in geriatric patients have been conducted. Phenytoin clearance tends to decrease with increasing age [see Clinical Pharmacology (12.3)]. Lower or less frequent dosing may be required [see Clinical Pharmacology (12.3) and Dosage and Administration (2.8)].
8.6 Renal and/or Hepatic Impairment, or Hypoalbuminemia
The liver is the site of biotransformation. Patients with impaired liver function, elderly patients, or those who are gravely ill may show early toxicity.
Because the fraction of unbound phenytoin (the active metabolite of fosphenytoin sodium injection) is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients.
After IV administration to patients with renal and/or hepatic disease, or in those with hypoalbuminemia, fosphenytoin clearance to phenytoin may be increased without a similar increase in phenytoin clearance. This has the potential to increase the frequency and severity of adverse events.
8.7 Use in Patients with Decreased CYP2C9 Function
Patients who are intermediate or poor metabolizers of CYP2C9 substrates (e.g., *1/*3, *2/*2, *3/*3) may exhibit increased phenytoin serum concentrations compared to patients who are normal metabolizers (e.g., *1/*1). Thus, patients who are known to be intermediate or poor metabolizers may ultimately require lower doses to maintain similar steady-state concentrations compared to normal metabolizers. In patients who are known to be carriers of the decreased function CYP2C9*2 or *3 alleles (intermediate and poor metabolizers), consider starting at the low end of the dosage range and monitor serum concentrations to maintain total phenytoin concentrations of 10 to 20 mcg/mL. If early signs of dose-related central nervous system (CNS) toxicity develop, serum concentrations should be checked immediately [see Clinical Pharmacology (12.5)].
10. Overdosage
Nausea, vomiting, lethargy, tachycardia, bradycardia, asystole, cardiac arrest, hypotension, syncope, hypocalcemia, metabolic acidosis, and death have been reported in cases of overdosage with fosphenytoin sodium injection.
Because fosphenytoin sodium injection is a prodrug of phenytoin, the following information about phenytoin overdosage may be helpful. Initial symptoms of acute phenytoin toxicity are nystagmus, ataxia, and dysarthria. Other signs include tremor, hyperreflexia, lethargy, slurred speech, nausea, vomiting, coma, and hypotension. Death is caused by respiratory and circulatory depression. The lethal dose of phenytoin in adults is estimated to be 2 to 5 grams. The lethal dose in pediatrics is not known.
There are marked variations among individuals with respect to serum phenytoin concentrations where toxicity occurs. Lateral gaze nystagmus usually appears at 20 µg/mL, ataxia at 30 µg/mL, and dysarthria and lethargy appear when the serum concentration is over 40 µg/mL. However, phenytoin concentrations as high as 50 µg/mL have been reported without evidence of toxicity. As much as 25 times the therapeutic phenytoin dose has been taken, resulting in serum phenytoin concentrations over 100 µg/mL, with complete recovery. Irreversible cerebellar dysfunction and atrophy have been reported after overdosage.
Formate and phosphate are metabolites of fosphenytoin sodium injection and therefore may contribute to signs of toxicity following overdosage. Signs of formate toxicity are similar to those of methanol toxicity and are associated with severe anion-gap metabolic acidosis. Large amounts of phosphate, delivered rapidly, could potentially cause hypocalcemia with paresthesia, muscle spasms, and seizures. Ionized free calcium levels can be measured and, if low, used to guide treatment.
Treatment: Treatment is nonspecific since there is no known antidote to fosphenytoin sodium injection or phenytoin overdosage.
The adequacy of the respiratory and circulatory systems should be carefully observed, and appropriate supportive measures employed. Hemodialysis can be considered since phenytoin (the active metabolite of fosphenytoin sodium injection) is not completely bound to plasma proteins. Total exchange transfusion has been used in the treatment of severe intoxication in children.
In acute overdosage the possibility of other CNS depressants, including alcohol, should be borne in mind.
11. Fosphenytoin Description
Fosphenytoin Sodium Injection, USP is a prodrug intended for parenteral administration; its active metabolite is phenytoin. 1.5 mg of fosphenytoin sodium is equivalent to 1 mg phenytoin sodium, and is referred to as 1 mg phenytoin sodium equivalents (PE). The amount and concentration of fosphenytoin is always expressed in terms of mg PE.
The pharmacological class of fosphenytoin sodium is hydantoin derivative, and the therapeutic class is anticonvulsant.
Fosphenytoin sodium injection is marketed in 2 mL vials containing a total of 100 mg PE/2 mL (50 mg PE/mL) and 10 mL vials containing a total of 500 mg PE/10 mL (50 mg PE/mL), for intravenous or intramuscular administration. The concentration of each vial is 50 mg PE/mL. Fosphenytoin sodium injection is supplied in vials as a ready-mixed solution in Water for Injection, USP, and Tromethamine, USP (TRIS), buffer adjusted to pH 8.6 to 9.0 with either Hydrochloric Acid, NF, or Sodium Hydroxide, NF. Fosphenytoin sodium injection is a clear, colorless to pale yellow, sterile solution.
The chemical name of fosphenytoin is 5,5-diphenyl-3-[(phosphonooxy)methyl]-2,4- imidazolidinedione disodium salt. The molecular structure of fosphenytoin is:
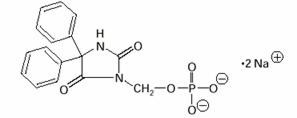
The molecular weight of fosphenytoin is 406.24.
12. Fosphenytoin - Clinical Pharmacology
12.1 Mechanism of Action
Fosphenytoin is a prodrug of phenytoin and accordingly, its anticonvulsant effects are attributable to phenytoin. The precise mechanism by which phenytoin exerts its therapeutic effect has not been established but is thought to involve the voltage-dependent blockade of membrane sodium channels resulting in a reduction in sustained high-frequency neuronal discharges.
12.3 Pharmacokinetics
Fosphenytoin
Absorption
Intravenous: When fosphenytoin sodium injection is administered by IV infusion, maximum plasma fosphenytoin concentrations are achieved at the end of the infusion.
Intramuscular: Fosphenytoin is completely bioavailable following IM administration of fosphenytoin sodium injection. Peak concentrations occur at approximately 30 minutes postdose. Plasma fosphenytoin concentrations following IM administration are lower but more sustained than those following IV administration due to the time required for absorption of fosphenytoin from the injection site.
Distribution
Fosphenytoin is extensively bound (95% to 99%) to human plasma proteins, primarily albumin. Binding to plasma proteins is saturable with the result that the percent bound decreases as total fosphenytoin concentrations increase. Fosphenytoin displaces phenytoin from protein binding sites. The volume of distribution of fosphenytoin increases with fosphenytoin sodium injection dose and rate, and ranges from 4.3 to 10.8 liters.
Elimination
The conversion half-life of fosphenytoin to phenytoin is approximately 15 minutes.
Metabolism
Following parenteral administration of fosphenytoin sodium injection, fosphenytoin is converted to the anticonvulsant phenytoin. The mechanism of fosphenytoin conversion has not been determined, but phosphatases probably play a major role. Fosphenytoin is metabolized to phenytoin, phosphate, and formate. For every mmol of fosphenytoin administered, one mmol of phenytoin is produced. The hydrolysis of fosphenytoin to phenytoin yields two metabolites, phosphate and formaldehyde. Formaldehyde is subsequently converted to formate, which is in turn metabolized via a folate dependent mechanism. Although phosphate and formaldehyde (formate) have potentially important biological effects, these effects typically occur at concentrations considerably in excess of those obtained when fosphenytoin sodium injection is administered under conditions of use recommended in this labeling.
Excretion
Fosphenytoin is not excreted in urine.
Phenytoin (after Fosphenytoin Sodium Injection administration)
In general, IM administration of fosphenytoin sodium injection generates systemic phenytoin concentrations that are similar enough to oral phenytoin sodium to allow essentially interchangeable use. The pharmacokinetics of fosphenytoin following IV administration of fosphenytoin sodium injection, however, are complex, and when used in an emergency setting (e.g., status epilepticus), differences in rate of availability of phenytoin could be critical. Studies have therefore empirically determined an infusion rate for fosphenytoin sodium injection that gives a rate and extent of phenytoin systemic availability similar to that of a 50 mg/min phenytoin sodium infusion. A dose of 15 to 20 mg PE/kg of fosphenytoin sodium injection infused at 100 to 150 mg PE/min yields plasma free phenytoin concentrations over time that approximate those achieved when an equivalent dose of phenytoin sodium (e.g., parenteral DILANTIN®) is administered at 50 mg/min [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)].
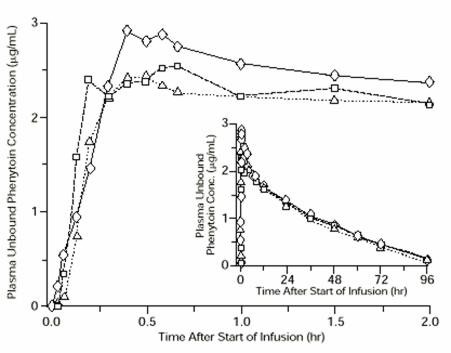
FIGURE 1. Mean plasma unbound phenytoin concentrations following IV administration of 1200 mg PE fosphenytoin sodium injection infused at 100 mg PE/min (triangles) or 150 mg PE/min (squares) and 1200 mg Dilantin infused at 50 mg/min (diamonds) to healthy subjects (N = 12). Inset shows time course for the entire 96-hour sampling period.
Following administration of single IV fosphenytoin sodium injection doses of 400 to 1200 mg PE, mean maximum total phenytoin concentrations increase in proportion to dose, but do not change appreciably with changes in infusion rate. In contrast, mean maximum unbound phenytoin concentrations increase with both dose and rate.
Absorption:
Fosphenytoin is completely converted to phenytoin following IV administration, with a half-life of approximately 15 minutes. Fosphenytoin is also completely converted to phenytoin following IM administration and plasma total phenytoin concentrations peak in approximately 3 hours.
Distribution:
Phenytoin is highly bound to plasma proteins, primarily albumin, although to a lesser extent than fosphenytoin. In the absence of fosphenytoin, approximately 12% of total plasma phenytoin is unbound over the clinically relevant concentration range. However, fosphenytoin displaces phenytoin from plasma protein binding sites. This increases the fraction of phenytoin unbound (up to 30% unbound) during the period required for conversion of fosphenytoin to phenytoin (approximately 0.5 to 1 hour postinfusion).
Elimination:
Mean total phenytoin half-life values (12.0 to 28.9 hr) following fosphenytoin sodium injection administration at these doses are similar to those after equal doses of parenteral Dilantin and tend to be greater at higher plasma phenytoin concentrations.
Metabolism
Phenytoin derived from administration of fosphenytoin sodium injection is extensively metabolized in the liver by the cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19. Phenytoin hepatic metabolism is saturable, and following administration of single IV fosphenytoin sodium injection doses of 400 to 1200 mg PE, total and unbound phenytoin AUC values increase disproportionately with dose.
Excretion
Phenytoin derived from administration of fosphenytoin sodium injection is excreted in urine primarily as 5-(p-hydroxyphenyl)-5-phenylhydantoin and its glucuronide; little unchanged phenytoin (1% to 5% of the fosphenytoin sodium injection dose) is recovered in urine.
Specific Populations
Age: Geriatric Population:
The effect of age on the pharmacokinetics of fosphenytoin was evaluated in patients 5 to 98 years of age. Patient age had no significant impact on fosphenytoin pharmacokinetics. Phenytoin clearance tends to decrease with increasing age (20% less in patients over 70 years of age relative to that in patients 20 to 30 years of age).
Sex/Race:
Gender and race have no significant impact on fosphenytoin or phenytoin pharmacokinetics.
Renal or Hepatic Impairment:
Increased fraction of unbound phenytoin (the active metabolite of fosphenytoin sodium injection) in patients with renal or hepatic disease, or in those with hypoalbuminemia has been reported.
Pregnancy:
It has been reported in the literature that the plasma clearance of phenytoin (the active metabolite of fosphenytoin sodium injection) generally increased during pregnancy, reached a peak in the third trimester and returned to the level of pre-pregnancy after few weeks or months of delivery [see Dosage and Administration (2.9)].
Drug Interaction Studies
Phenytoin derived from administration of fosphenytoin sodium injection is extensively metabolized in the liver by the cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19 [see Drug Interactions (7.1, 7.2)]. No drugs are known to interfere with the conversion of fosphenytoin to phenytoin. Conversion could be affected by alterations in the level of phosphatase activity, but given the abundance and wide distribution of phosphatases in the body it is unlikely that drugs would affect this activity enough to affect conversion of fosphenytoin to phenytoin.
The pharmacokinetics and protein binding of fosphenytoin, phenytoin, and diazepam were not altered when diazepam and fosphenytoin sodium injection were concurrently administered in single submaximal doses.
12.5 Pharmacogenomics
CYP2C9 activity is decreased in individuals with genetic variants such as the CYP2C9*2 and CYP2C9*3 alleles. Carriers of variant alleles, resulting in intermediate (e.g., *1/*3, *2/*2) or poor metabolism (e.g., *2/*3, *3/*3) have decreased clearance of phenytoin. Other decreased or nonfunctional CYP2C9 alleles may also result in decreased clearance of phenytoin (e.g., *5, *6, *8, *11).
The prevalence of the CYP2C9 poor metabolizer phenotype is approximately 2–3% in the White population, 0.5–4% in the Asian population, and <1% in the African American population. The CYP2C9 intermediate phenotype prevalence is approximately 35% in the White population, 24% in the African American population, and 15–36% in the Asian population [see Warnings and Precautions (5.4) and Use in Specific Populations (8.7)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis [see Warnings and Precautions (5.9)]
The carcinogenic potential of fosphenytoin has not been assessed. In carcinogenicity studies, phenytoin (active metabolite of fosphenytoin) was administered in the diet to mice (10, 25, or 45 mg/kg/day) and rats (25, 50, or 100 mg/kg/day) for 2 years. The incidences of hepatocellular tumors were increased in male and female mice at the highest dose. No increases in tumor incidence were observed in rats. The highest doses tested in these studies were associated with peak plasma phenytoin levels below human therapeutic concentrations.
In carcinogenicity studies reported in the literature, phenytoin was administered in the diet for 2 years at doses up to 600 ppm (approximately 160 mg/kg/day) to mice and up to 2400 ppm (approximately 120 mg/kg/day) to rats. The incidences of hepatocellular tumors were increased in female mice at all but the lowest dose tested. No increases in tumor incidence were observed in rats.
Mutagenesis
An increase in structural chromosome aberrations were observed in cultured V79 Chinese hamster lung cells exposed to fosphenytoin in the presence of metabolic activation. No evidence of mutagenicity was observed in bacteria (Ames test) or Chinese hamster lung cells in vitro, and no evidence for clastogenic activity was observed in an in vivo mouse bone marrow micronucleus assay.
Impairment of Fertility
Fosphenytoin was administered to male and female rats during mating and continuing in females throughout gestation and lactation at doses of 50 mg PE/kg or higher. No effects on fertility were observed in males. In females, altered estrous cycles, delayed mating, prolonged gestation length, and developmental toxicity were observed at all doses, which were associated with maternal toxicity. The lowest dose tested is approximately 40% of the maximum human loading dose on a mg/m2 basis.
14. Clinical Studies
Infusion tolerance was evaluated in clinical studies. One double-blind study assessed infusion-site tolerance of equivalent loading doses (15 to 20 mg PE/kg) of fosphenytoin sodium injection infused at 150 mg PE/min or phenytoin infused at 50 mg/min. The study demonstrated better local tolerance (pain and burning at the infusion site), fewer disruptions of the infusion, and a shorter infusion period for fosphenytoin sodium injection-treated patients (Table 8).
TABLE 8. Infusion Tolerance of Equivalent Loading Doses of IV Fosphenytoin Sodium Injection and IV Phenytoin
|
IV Fosphenytoin Sodium Injection N=90 |
IV Phenytoin N=22 |
|
|
Local Intolerance Infusion Disrupted Average Infusion Time |
9%a 21% 13 min |
90% 67% 44 min |
aPercent of patients
Fosphenytoin sodium injection-treated patients, however, experienced more systemic sensory disturbances [see Warnings and Precautions (5.10)]. Infusion disruptions in fosphenytoin sodium injection-treated patients were primarily due to systemic burning, pruritus, and/or paresthesia while those in phenytoin-treated patients were primarily due to pain and burning at the infusion site (see Table 8). In a double-blind study investigating temporary substitution of fosphenytoin sodium injection for oral phenytoin, IM fosphenytoin sodium injection was as well-tolerated as IM placebo. IM fosphenytoin sodium injection resulted in a slight increase in transient, mild to moderate local itching (23% of fosphenytoin sodium injection-treated patients vs. 11% of IM placebo-treated patients at any time during the study). This study also demonstrated that equimolar doses of IM fosphenytoin sodium injection may be substituted for oral phenytoin sodium with no dosage adjustments needed when initiating IM or returning to oral therapy. In contrast, switching between IM and oral phenytoin requires dosage adjustments because of slow and erratic phenytoin absorption from muscle.
16. How is Fosphenytoin supplied
16.1 How Supplied
Fosphenytoin Sodium Injection, USP is a clear, colorless to pale yellow solution supplied as follows:
|
mg phenytoin sodium equivalents (PE) per vial |
Volume per vial |
Package Configuration |
NDC |
|
500 mg PE/10 mL vial |
10 mL per vial |
Package contains 10 vials |
NDC 0641-6137-10 |
|
100 mg PE/2 mL vial |
2 mL per vial |
Package contains 25 vials |
NDC 0641-6136-25 |
Both sizes of vials contain Tromethamine, USP (TRIS), Hydrochloric Acid, NF, or Sodium Hydroxide, NF, and Water for Injection, USP.
Fosphenytoin sodium injection should always be prescribed in phenytoin sodium equivalents (PE) [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)].
1.5 mg of fosphenytoin sodium is equivalent to 1 mg phenytoin sodium, and is referred to as 1 mg PE. The amount and concentration of fosphenytoin is always expressed in terms of mg of phenytoin sodium equivalents (PE). Fosphenytoin’s weight is expressed as phenytoin sodium equivalents to avoid the need to perform molecular weight-based adjustments when substituting fosphenytoin for phenytoin or vice versa.
16.2 Storage and Handling
Store under refrigeration at 2°C to 8°C (36°F to 46°F). The product should not be stored at room temperature for more than 48 hours. Vials that develop particulate matter should not be used.
Vials are single-dose only. After opening, any unused product should be discarded.
Not made with natural rubber latex.
Brands listed are the trademarks of their respective owners.
17. Patient Counseling Information
Cardiovascular Risk Associated with Rapid Infusion
Inform patients that rapid intravenous administration of fosphenytoin sodium injection increases the risk of adverse cardiovascular reactions, including
severe hypotension and cardiac arrhythmias. Cardiac arrhythmias have included bradycardia, heart block, ventricular tachycardia, and ventricular fibrillation which have resulted in asystole, cardiac arrest, and death. Patients should report cardiac signs or symptoms to their healthcare provider [see Warnings and Precautions (5.2)].
Withdrawal of Antiepileptic Drugs
Advise patients not to discontinue use of fosphenytoin sodium injection without consulting with their healthcare provider. Fosphenytoin sodium injection should normally be gradually withdrawn to reduce the potential for increased seizure frequency and status epilepticus [see Warnings and Precautions (5.3)].
Serious Dermatologic Reactions
Advise patients of the early signs and symptoms of severe cutaneous adverse reactions and to report any occurrence immediately to a physician [see Warnings and Precautions (5.4)].
Potential Signs of Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) and Other Systemic Reactions
Advise patients of the early toxic signs and symptoms of potential hematologic, dermatologic, hypersensitivity, or hepatic reactions. These symptoms may include, but are not limited to, fever, sore throat, rash, ulcers in the mouth, easy bruising, lymphadenopathy, facial swelling, and petechial or purpuric hemorrhage, and in the case of liver reactions, anorexia, nausea/vomiting, or jaundice. Advise the patient that, because these signs and symptoms may signal a serious reaction, that they must report any occurrence immediately to a physician. In addition, advise the patient that these signs and symptoms should be reported even if mild or when occurring after extended use [see Warnings and Precautions (5.4, 5.5, 5.6, 5.8, 5.9)].
Angioedema
Advise patients to discontinue fosphenytoin sodium injection and seek immediate medical care if they develop signs or symptoms of angioedema such as facial, perioral, or upper airway swelling [see Warnings and Precautions (5.7)].
Hyperglycemia
Advise patients that fosphenytoin sodium injection may cause an increase in blood glucose levels [see Warnings and Precautions (5.16)].
Effects of Alcohol Use and Other Drugs and Over-the-Counter Drug Interactions
Caution patients against the use of other drugs or alcoholic beverages without first seeking their physician's advice [see Drug Interactions (7.1, 7.2)].
Inform patients that certain over-the-counter medications (e.g., cimetidine and omeprazole), vitamins (e.g., folic acid), and herbal supplements (e.g., St. John's wort) can alter their phenytoin levels.
Use in Pregnancy
Inform pregnant women and women of childbearing potential that use of fosphenytoin sodium injection during pregnancy can cause fetal harm, including an increased risk for cleft lip and/or cleft palate (oral clefts), cardiac defects, dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits. When appropriate, counsel pregnant women and women of childbearing potential about alternative therapeutic options. Advise women of childbearing potential who are not planning a pregnancy to use effective contraception while using fosphenytoin sodium injection, keeping in mind that there is a potential for decreased hormonal contraceptive efficacy [see Drug Interactions (7.2)].
Instruct patients to notify their physician if they become pregnant or intend to become pregnant during therapy, and to notify their physician if they are breastfeeding or intend to breastfeed during therapy [see Use in Specific Populations (8.1, 8.2)].
Encourage patients to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy [see Use in Specific Populations (8.1)].
Manufactured by:
Hikma Pharmaceuticals USA Inc.
Berkeley Heights, NJ 07922
462-438-15
Revised January 2023
PRINCIPAL DISPLAY PANEL
NDC 0641-6136-01 Rx only
Fosphenytoin
Sodium Injection, USP
100 mg PE per 2 mL
(50 mg PE/mL)
(PE = phenytoin
sodium equivalents)
For IM or IV use
2 mL Vial

NDC 0641-6136-25 Rx only
Fosphenytoin
Sodium Injection, USP
100 mg PE per 2 mL
(50 mg PE/mL)
(PE = phenytoin sodium equivalents)
For IM or IV use
25 x 2 mL Single Dose Vials

PRINCIPAL DISPLAY PANEL
NDC 0641-6137-01 Rx only
Fosphenytoin
Sodium Injection, USP
500 mg PE per 10 mL
(50 mg PE/mL)
(PE = phenytoin sodium equivalents)
For IM or IV use
10 mL Vial
NDC 0641-6137-10 Rx only
Fosphenytoin
Sodium Injection, USP
500 mg PE in 10 mL
(50 mg PE/mL)
(PE = phenytoin sodium equivalents)
For IM or IV use
10 x 10 mL Single Dose Vials

| FOSPHENYTOIN SODIUM
fosphenytoin sodium injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FOSPHENYTOIN SODIUM
fosphenytoin sodium injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Hikma Pharmaceuticals USA Inc. (946499746) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Hikma Pharmaceuticals USA Inc. | 946499746 | analysis(0641-6136, 0641-6137) , label(0641-6136, 0641-6137) , manufacture(0641-6136, 0641-6137) | |
More about fosphenytoin
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Drug class: hydantoin anticonvulsants
- Breastfeeding
- En español